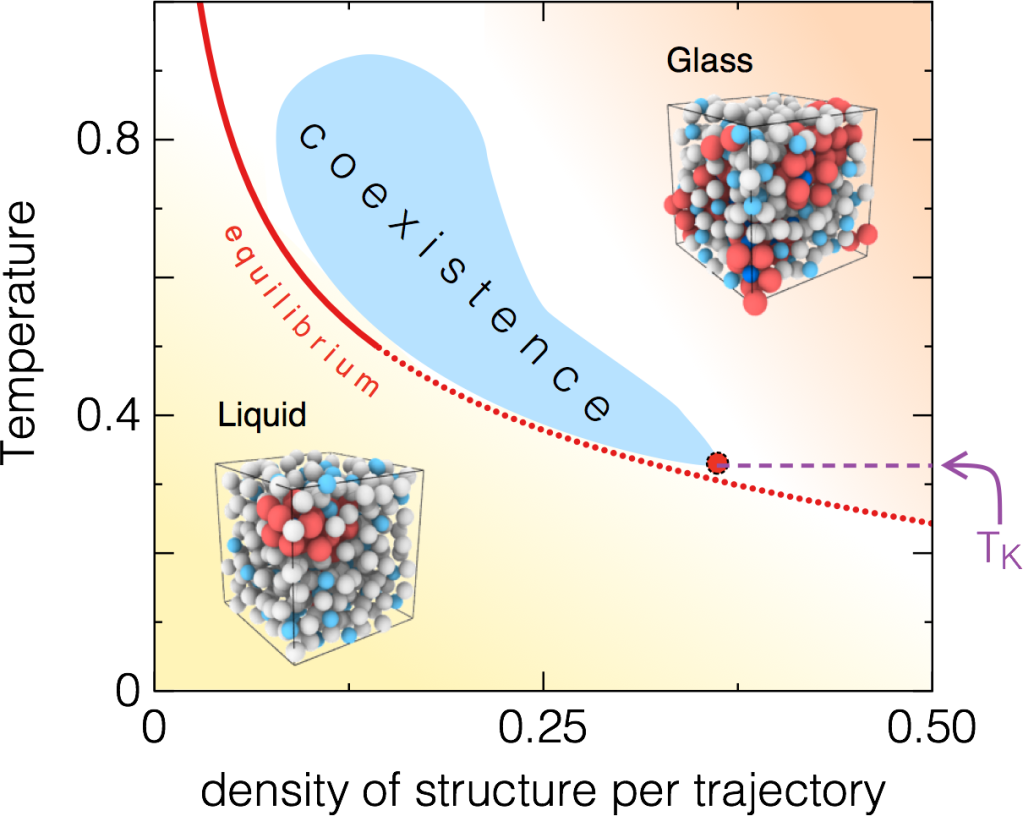
Complex Disordered Systems
Arrested states: Glasses
Energy landscapes
- A system of many partcles has many degrees of freedom.
- The free energy is a scalar function of all these degrees of freedom: F(x_1, x_2, \ldots, x_N).
- We can think of it is the altitude of a landscape in a high-dimensional space.
- The landscape represents the (negative log) of the probability distribution of configurations: low energy = high probability.
- it depends on the intensive parameters (temperature, pressure, etc..)
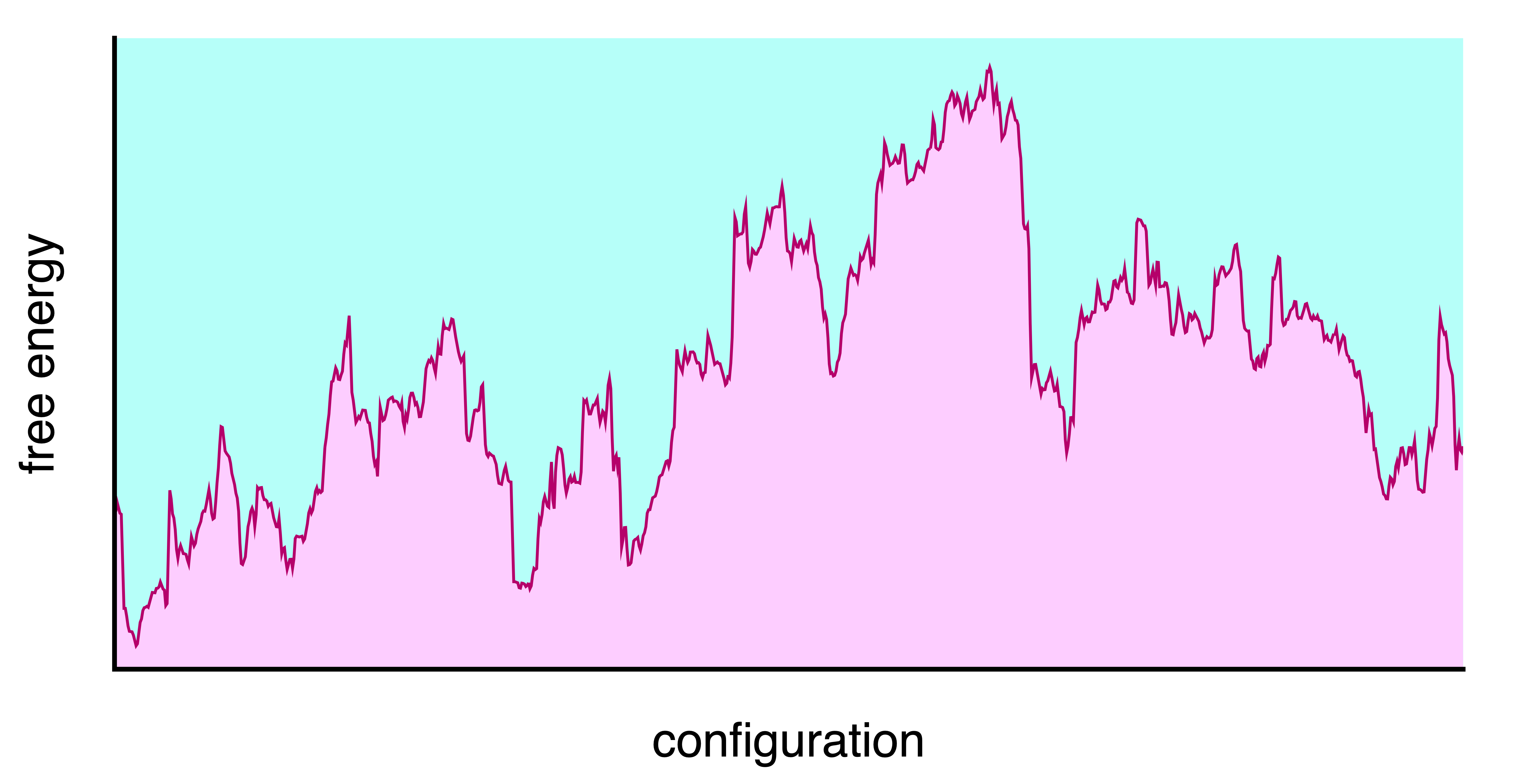
Free energy landscape of a complex system at some temperature T and pressure P
Energy landscapes
- Different minima correspond to different conformations or states of the system. (e.g. liquid/gas, crystal/amorphous solid, folded/unfolded protein)
- They are separated by energy barriers \Delta F
- The time to transition is often modelled as Arrhenius \tau \sim \exp\left(\dfrac{\Delta F}{k_{B} T}\right)
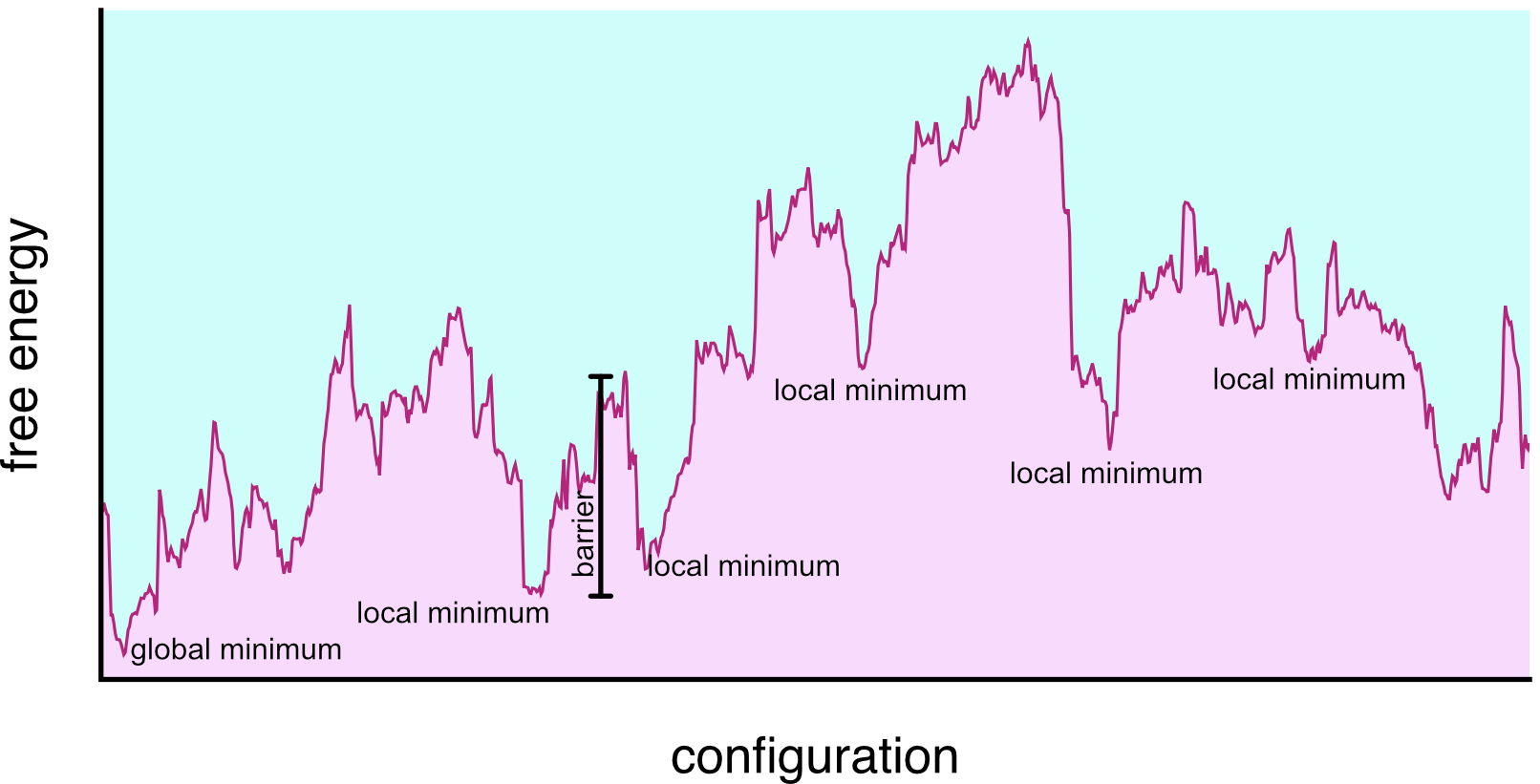
Minima in the free energy landscape
Metabasins
When the temperature allows it, connected minima can be grouped into single basins called metabasins.
- they are not just energy minima, they include entropy
- they can behave as bone-fide local equilibria
- These are akin to the metastable states.
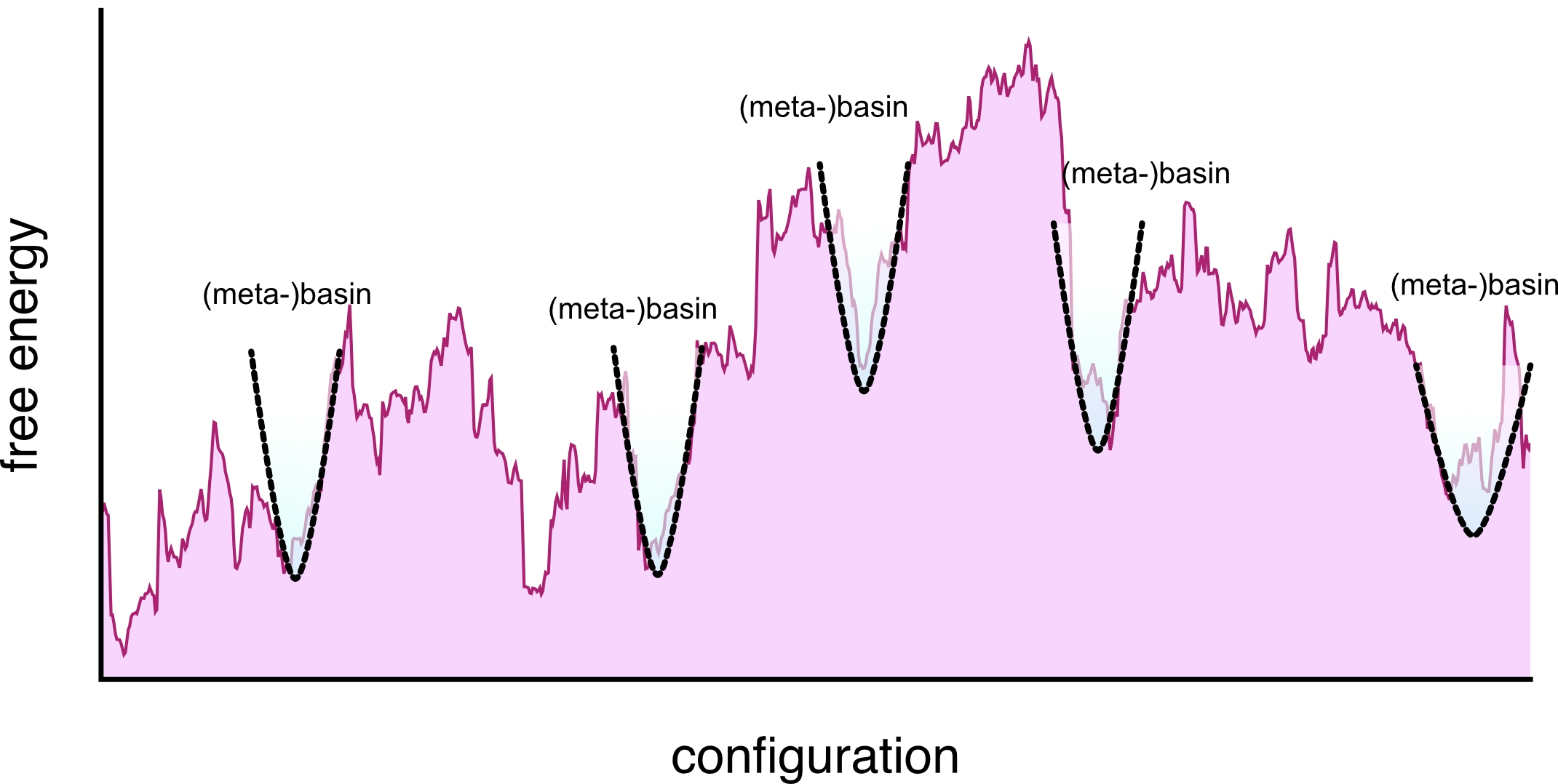
metabasins in the free energy landscape
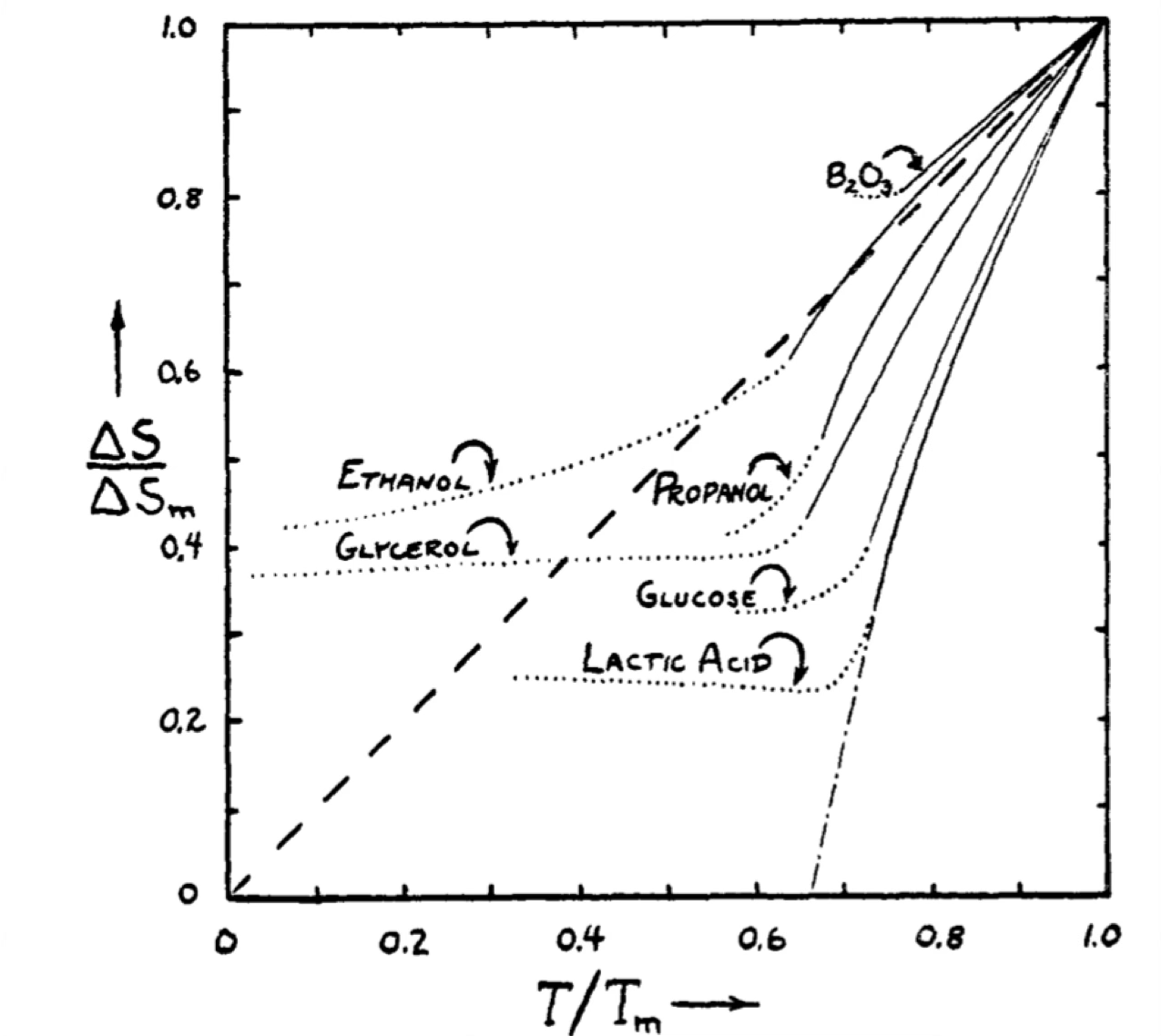
Configurational entropy
As we take liquids to low temperatures, we can separate the total entropy into a vibrational and configurational part and define S_{conf}(T) = S(T) -S_{vib}(T)
For several fluids, this decreases with temperature in an intriguing way.
[…] Perhaps in some instances a thermodynamic “freezing-in” of degrees of freedom does take place as a desperate result of the liquid’s excessive generosity with its limited supply of entropy and energy as its temperature is lowered below the melting point. This would imply the existence of some kind of state of high order for the liquid at low temperature which differs from the normal crystalline state. A plausible structure for such a state seems, however, difficult to conceive, and we believe that the paradox is better resolved in another way […] (W. Kauzmann, Chem. Rev. 43, 219 (1948)).
Glassy systems
But glassy physics is broader than glassy materials: dense assemblies in general will display glassy dynamics.

(a) Colloidal glasses, (b) dense cellular tissues, (c) ant colonies from Facets fo glassy physics, Berthier and Ediger, Physics Today (2016)
Glass formation
To form a glass we:
- start from the liquid at high temperature
- cool with a finite cooling rate fast enough to avoid crystallisation
- enter a local equilibrium state (the supercooled liquid)
- at some point the relaxation time \tau becomes longer than the experimental time scale and the system falls out of equilibrium into a glassy state
- changing the cooling rate changes the glass transition temperature T_g and the final glass properties

Macroscopic Glass properties
- Glasses are solids: they have a finite shear modulus and yield stress
- Glasses are amorphous: they lack long-range order (no Bragg peaks in scattering experiments)
- Glasses are history-dependent: their properties depend on the cooling rate and thermal history
- Glasses exhibit ageing: their properties slowly evolve with time as they explore deeper minima in the energy landscape
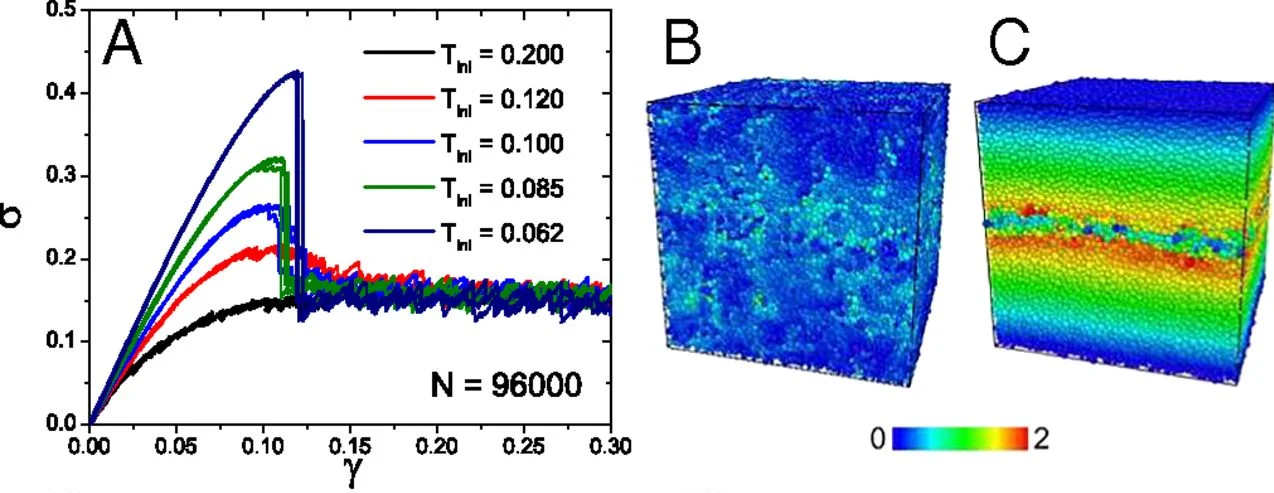
Example of brittle response in a simulated glassy model form Ozawa et al PNAS (2018): (a) stress-strain curves for different preparation temperatures; (b) initial deformation field, (b) final deformation field demonstrating fracture
Slowing down
Their solid-like behavior arises from the dramatic slowing down of their dynamics as they approach the glass transition.
We quantify this with density autocorrelation functions such as the intermediate scattering function: F(k, t) = \frac{1}{N} \left\langle \sum_{j=1}^{N} e^{i \mathbf{k} \cdot [\mathbf{r}_j(t) - \mathbf{r}_j(0)]} \right\rangle
It decays in two steps:
- a fast decay corresponding to vibrations within the cage formed by neighbouring particles, occuring on a timescale called the beta relaxation time \tau_{\beta}
- a slow decay corresponding to structural relaxation as particles escape their cages, taking place on the much longer alpha relaxation time \tau_{\alpha}
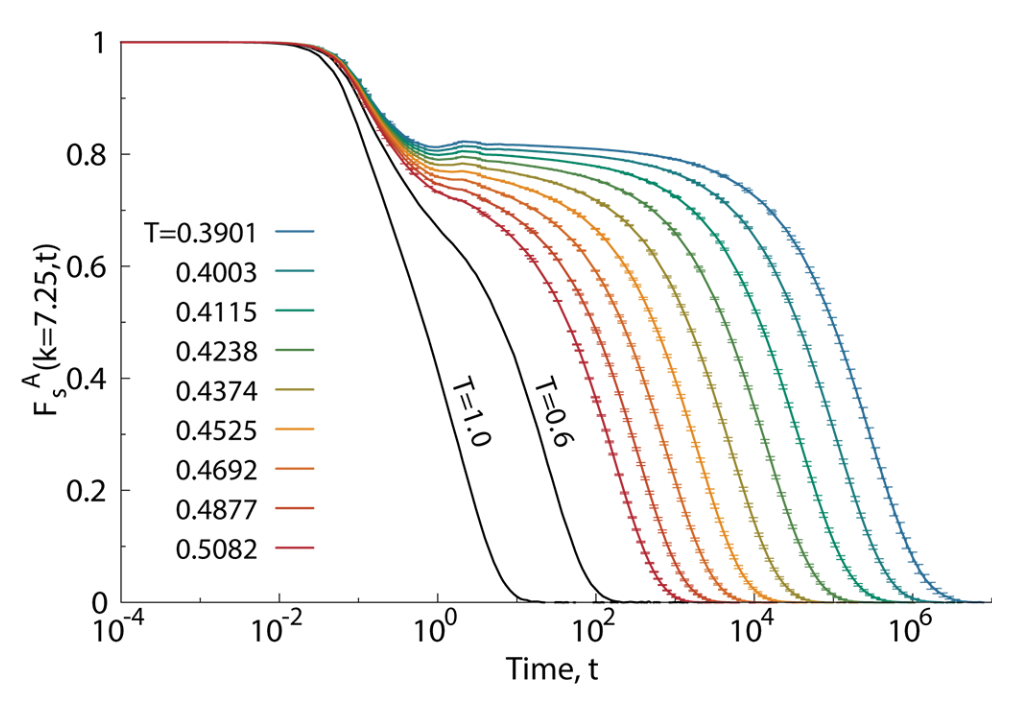
Divergence of relaxation time and viscosity
The alpha relaxation time \tau_{\alpha} is proportional to the viscosity \eta and both increase dramatically as the temperature approaches the glass transition temperature T_g.
Phenomenologically one distinguishes between:
- Strong glassformers: Arrhenius behaviour of \tau_{\alpha} and \eta \sim \exp\left(\dfrac{E_a}{k_{B} T}\right)
- Fragile glassformers: super-Arrhenius behaviour \tau_{\alpha} and \eta. Often modelled with the Vogel-Fulcher-Tammann (VFT) equation \tau \sim \exp\left(\dfrac{A}{T - T_0}\right) where T_0 corrsponds to a divergence at finite temperature \to phase transition?

A thermodynamic theory: the Adam-Gibbs scenario
The Adam-Gibbs model links the dramatic slowdown of dynamics to the underlying thermodynamics through configurational entropy S_{\rm conf}.
- The key idea is the existense of cooperatively rearranging regions (CRRs): to relax, particles need to move together and explore a new metabasin in the energy landscape.
Mosaic of M_{CRR} independent cooperatively rearranging regions of size n_{CRR} particles each. The total number of particles is N = M_{CRR} n_{CRR}.
We assume that the total configurational entropy is additive and that each CRR contributes a constant amount s_{conf}, irrespective of its size
S_{conf} = M_{CRR} s^* = \dfrac{N}{n_{CRR}} s^*
Rearranging gives the size of a CRR as a function of the configurational entropy per particle S_{conf}/N: n_{CRR}(T) \propto \dfrac{1}{S_{conf}(T)/N}

A dynamic theory: dynamical facilitation
Alternative model: glassy systems are universally heterogeneous in space and time: their mobility is very broadly distributed, with regions of high mobility coexisting with regions of low mobility.
This dynamical coexistence is called dynamical heterogeneity and can be formalised as a dynamical phase transition.
Key idea: Mobility facilitates more mobility
- Relaxation occurs only near already-mobile regions
- No underlying thermodynamic transition required
Temperature dependence:
- High T: Many mobile regions → fast relaxation
- Low T: Few mobile regions → cooperative motion required → slow relaxation
The relaxation time follows a parabolic law \log \tau_\alpha(T) \sim J^2 \left( \frac{1}{T} - \frac{1}{T_0} \right)^2
Unlike VFT, no finite-temperature divergence, no criticality, but still captures super-Arrhenius behavior.
 - Recent evidence suggests facilitation as a dominating mechanism for relaxation at low temperatures.
- Recent evidence suggests facilitation as a dominating mechanism for relaxation at low temperatures.
Unification
- Much more to glassy dynamics:
- geometrical frustration
- local structures
- growth of lengthscales
- jamming
- mean field solutions (see Nobel Prize Giorgio Parisi)
Ongoing work to unify the various scenarios.
- Example: Turci et al PRX (2017): a numerical demonstration that an extension dynamical facilitation is compatible with decreasing entropies, emergence of local structures and criticality.