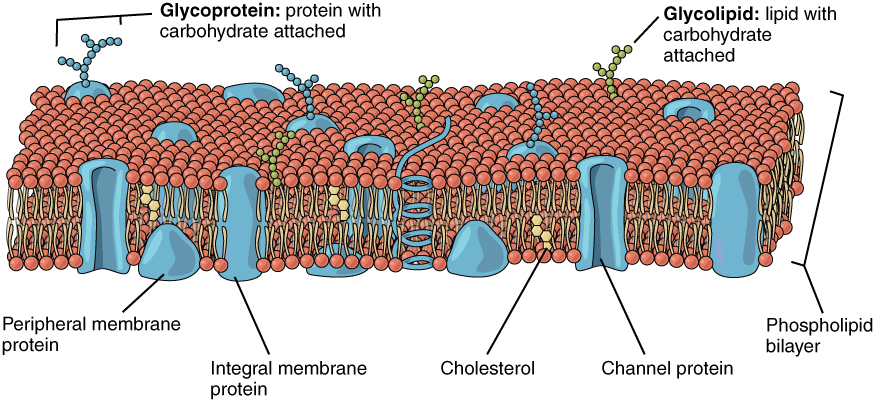
Water is the standard solvent for a lot of chemistry and (almost all) biology.
Molecules in water can be classified by their affinity to water:
Hydrophilic (“water-loving”) molecules interact favorably with water (e.g., via hydrogen bonding or dipole interactions).
Hydrophobic (“water-fearing”) molecules do not interact favorably with water (e.g., non-polar molecules).
Amphiphiles are molecules that contain both hydrophilic and hydrophobic parts. They include:
Surfactants (detergents, soaps, SURFace ACTive AgeNT)
Lipids (cell membranes)
Some proteins (with hydrophobic and hydrophilic amino acids)
Schematic model of a surfactant
Hydrophobicity and amphiphiles
Phospholipid bilayers form the cellular membranes. They self-assemble!
Hydrophobicity in brief
The hydrophobic interactions is a topic of active research.
Various approaches
(from to 1950s) structural approach: water forms cages around molecules. When the cage is disrupted, entropy increases. Solutes come together to minimise the disruption.
(from 2000s) interfaces approach: water is a fluctuating medium. Large hydrophobic solutes create interfaces that suppress fluctuations. Solutes come together to minimise the interface area.
(current research) avoided criticality: water is close to a liquid-vapour critical point. Local curvature and interactions promote large density fluctuations. Solutes interact on scales controlled by these fluctuations.
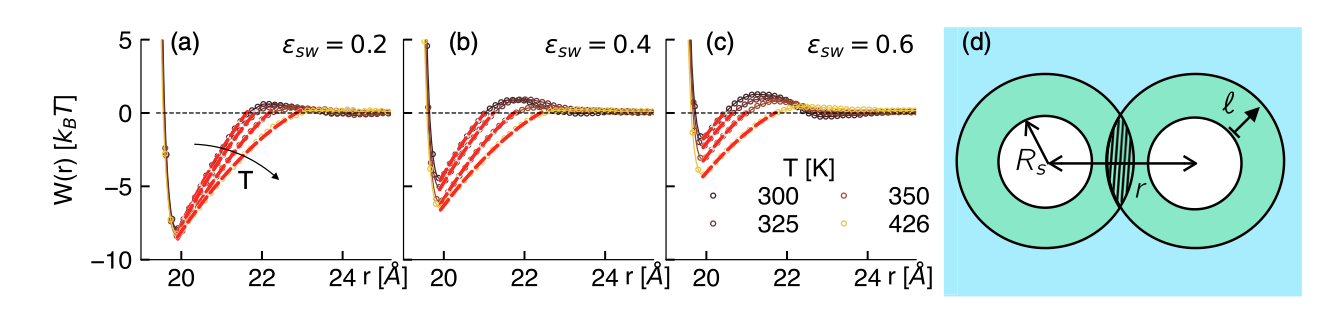
Effective hydrophobic interactions between spherical solutes of variable solute-solvent affinities \varepsilon_{sw}. A depletion-like construction produces a surface tension-controlled interaction. from Wilding & Turci PRR (2025)
In all cases, the hydrophobic effect originates in fluctuations of the solvent, i.e. the way the solvent explores the available configuration.
Self assembly of surfactants
viewof temperature = Inputs.range([0.01,2], {step:0.05,label:"Temperature (kT)",value:0.5})
viewof numChains = Inputs.range([3,300], {step:10,label:"Number of Chains",value:100})
Code
// This is ObservableJS code // You can run at observableehq.com or convert it to an equivalent (and faster?) Python version if you likeviewof simulation = {// --- Parameters ---const width =40, height =40;const chainLength =3;const chainsToCreate = numChains;const T = temperature;// const numChains = 10;const E_TS =1.0;// tail-solvent energy (solvophobic)const E_HS =-1.5;// head-solvent energy (solvophilic)// const T = 0.1; // temperature (kT units)// --- Initialize grid and chains ---let grid =Array.from({length: width}, () =>Array(height).fill(null));let chains = [];// Periodic boundary helperfunctionmod(n, m) { return ((n % m) + m) % m; }functiongetNeighbors(x, y) {return [ {x:mod(x -1, width),y: y}, {x:mod(x +1, width),y: y}, {x: x,y:mod(y -1, height)}, {x: x,y:mod(y +1, height)} ]; }functionplaceChains() {for (let id =0; id < numChains; id++) {for (let tries =0; tries <100; tries++) {let x =Math.floor(Math.random() * width);let y =Math.floor(Math.random() * height);if (grid[x][y]) continue;let chain = [{x, y,type:'head'}]; grid[x][y] = {id,type:'head'};let ok =true;for (let i =1; i < chainLength; i++) {let last = chain[chain.length-1];let nbs =getNeighbors(last.x, last.y).filter(p =>!grid[p.x][p.y]);if (nbs.length===0) { ok =false;break; }let next = nbs[Math.floor(Math.random() * nbs.length)]; chain.push({...next,type:'tail'}); grid[next.x][next.y] = {id,type:'tail'}; }if (ok) { chains.push({id,segments: chain});break; }else { chain.forEach(p => grid[p.x][p.y] =null); } } } }functionenergy(seg) {let solventNbs =getNeighbors(seg.x, seg.y).filter(p =>!grid[p.x][p.y]).length;return seg.type==='head'? solventNbs * E_HS : solventNbs * E_TS; }functionattemptMove(chain) {const forward =Math.random() <0.5;const tail = forward ? chain.segments[0] : chain.segments.at(-1);const head = forward ? chain.segments.at(-1) : chain.segments[0];const options =getNeighbors(head.x, head.y).filter(p =>!grid[p.x][p.y]);if (options.length===0) return;const next = options[Math.floor(Math.random() * options.length)];const dE_old =energy(tail); grid[tail.x][tail.y] =null;const dE_new =energy({...next,type: tail.type}); grid[tail.x][tail.y] = {id: chain.id,type: tail.type};const deltaU = dE_new - dE_old;const accept = deltaU <0||Math.random() <Math.exp(-deltaU / T);if (accept) { grid[tail.x][tail.y] =null; grid[next.x][next.y] = {id: chain.id,type: tail.type};if (forward) { chain.segments.shift(); chain.segments.push({...next,type: tail.type}); } else { chain.segments.pop(); chain.segments.unshift({...next,type: tail.type}); } } }// --- Visualization ---const svg = d3.create("svg").attr("viewBox",`0 0 ${width}${height}`).style("width","400px").style("height","400px").style("border","1px solid #ccc");const g = svg.append("g");functiondraw() {const data = chains.flatMap(c => c.segments); g.selectAll("circle").data(data, d =>`${d.x}-${d.y}`).join("circle").attr("cx", d => d.x+0.5).attr("cy", d => d.y+0.5).attr("r",0.45).attr("fill", d => d.type==="head"?"blue":"red"); }// --- Main Loop ---placeChains();draw();let running =true;const button =html`<button>⏸ Pause</button>`; button.onclick= () => { running =!running; button.textContent= running ?"⏸ Pause":"▶ Resume"; }; (async () => {while (true) {if (running) {for (let i =0; i < chains.length; i++) {const c = chains[Math.floor(Math.random() * chains.length)];attemptMove(c); }draw(); }awaitnewPromise(r =>setTimeout(r,50)); } })();returnhtml`<div>${button}<br>${svg.node()}</div>`;}
Self assembly of surfactants
Surfactants are at the molecular scale so they are nanometer sized
Diffusion is fast (e.g. use D=\frac{k_B T}{6 \pi \eta R} to compare a colloid to a surfactant molecule)
\to quickly explore possible conformations and minimise free energy.
The amphiphilic structure means that special orientations can be achieved to satisfy the energetic constraints.
The molecules spontaneously form structures: this is called self-assembly.
Underlying phase transitions typically drive the self-assembly.
Aggregation: general case
Consider a generic problem of aggregation of particles (generic)
Simple model: we have cluster of size N and a free energy cost \epsilon_N pto add a particle to the cluster.
Let \mu_1 and \mu_N be the chemical potential of isolated particles and aggregates of size N respectively.
In equilibrium, \mu_1 = \mu_N := \mu
We can express \mu in terms of
the interaction energy from being in the aggregate \epsilon_N
the entropy of the aggregate as a whole \propto {\text{ number of aggregates}}\times \ln {\text{ number of aggregates}}
Aggregation: general case
Let the overall volume fraction to be \phi
Call X_N the volume fraction in an aggregate of size N, so that \sum_N X_N = \phi.
The numbers of aggregates of size N per unit volume is then simply X_N/N
Example
Suppose \phi = 0.1 (10% of the volume is particles).
which is a monotonically decreasing function of N.
Define \alpha k_B T = \gamma v^{2/3} and extract a relation between X_N and X_1 parametrised solely by \alpha, i.e.
\text{number of aggregates of size N per unit volume} = \dfrac{X_N}{N}\sim (X_1 e^{\alpha})^N
Aggregation: general case
\text{number of aggregates of size N per unit volume} = \dfrac{X_N}{N}\sim (X_1 e^{\alpha})^N This presents us with a few cases depending on \alpha= \gamma v^{2/3}/k_B T
X_1 e^{\alpha} < 1: Few isolated particles, and (X_1 e^{\alpha})^N becomes vanishingly small for large N
X_1 e^{\alpha} = 1 (critical point): Aggregates of all sizes become equally probable. The system undergoes phase separation into a dilute phase of isolated monomers (with X_1 pinned at e^{-\alpha}) in coexistence with a dense phase of very large aggregates.
X_1 e^{\alpha} > 1: Above the critical aggregation concentration, the system cannot remain homogeneous and phase separates. The volume fraction \phi at which this occurs is called critical aggregation concentration, or CAC.
Earlier we assumed that the free energy per particle was \epsilon_N = \epsilon_\infty +\gamma\left(\dfrac{v^2}{N}\right)^{1/3} a monotonically decreasing function of N.
For surfactants, it is not necessarily the case.
Typically \epsilon_N has a minimum at some finite N=N^* resulting from the fact that beyond a certain size, the aggregate becomes unfavourable (e.g., due to packing constraints, curvature energy, etc).
Therefore the distribution of aggregates of size N is peaked around N^*.
This leads to the formation of micelles: aggregates of a characteristic size.
Aggregation: surfactants case
Surfactants in solution: normally, this is done in water in the presence of an interface with air. Due to their nature as amphiphiles, the surfactants typically sit at the air-water interface in a dynamical, equilibrium process that exchanges monomers between the bulk and the surface. The bulk surfactants, when the concentration is larger than a critical value, also self-assemble into micellar structures, which are at equilibrium with the isolated single surfactants.
Critical micellar concentration (CMC)
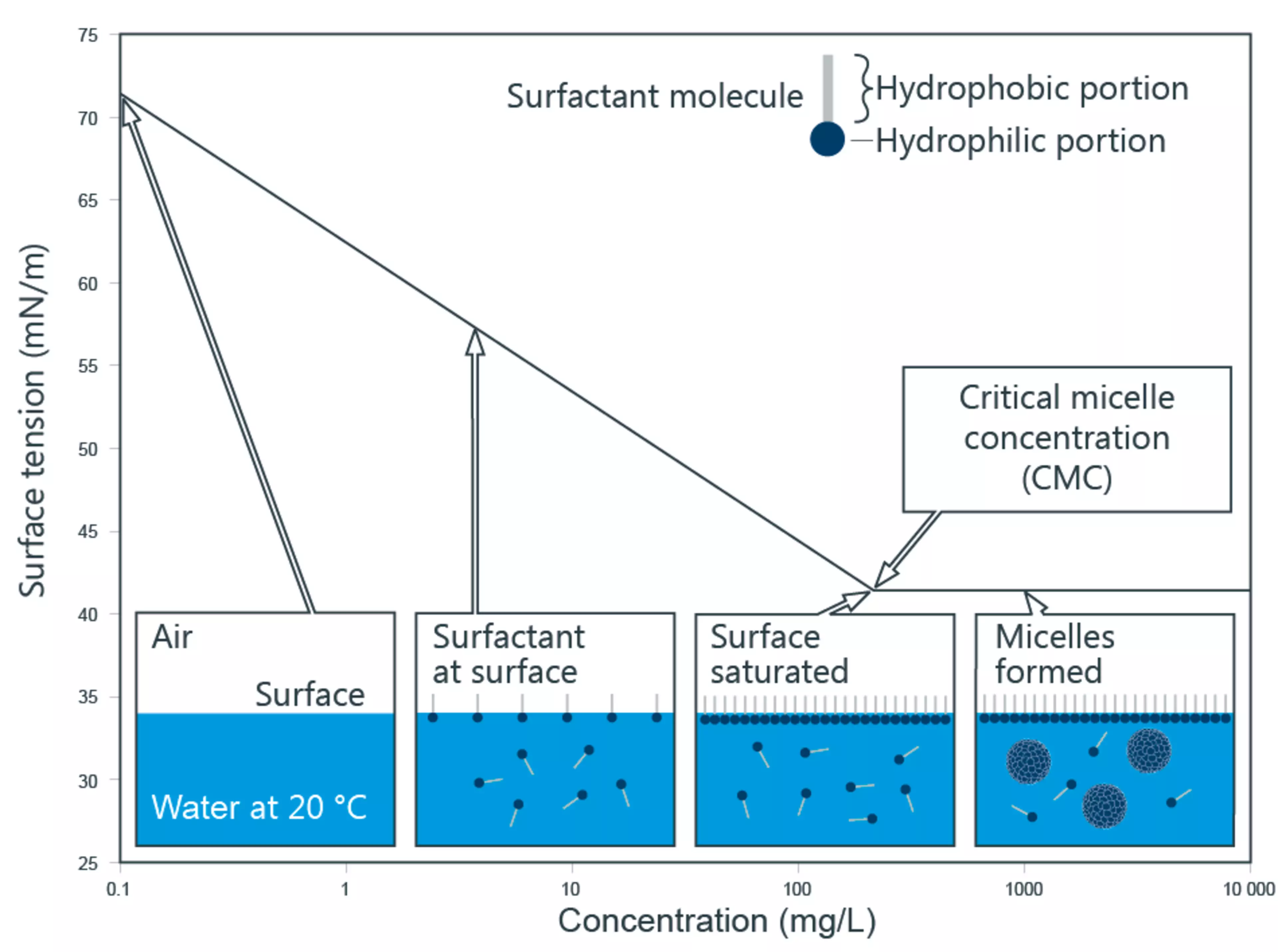
The addition of surfactants to water initially reduces the water-air surface tension, but when the surface is saturated, further addition leads to crossing the a critical micellar concentration (CMC) and micelle formation.
Below the CMC, almost all surfactant molecules are present as monomers
Above the CMC, additional surfactant molecules predominantly form micelles of size N^* while the monomer concentration remains nearly constant.
The aggregation in water follows specific patterns:
hydrophobic tails cluster together to avoid water
hydrophilic heads face the water
The structure on the micelle surface (formed by the heads) is disordered and fluid-like, similar to a 2D liquid.
Surface tension of a surfactant solution with increasing concentration, formation of micelles, from Kruss Scientific
Shape of surfactant assemblies
Geometric model of a surfactant molecule:
optimal headgroup area a_{0}: depends on chemical structure, pH and temeprature.
volume v of the hydrophobic part: The hydrophobic part usually consists of hydrocarbon chains. Its volume v\propto n the number of carbon atoms in the chain.
critical chain length\boldsymbol{l}_{\boldsymbol{c}} : the effective length of the chain, taking into account chain flexibility, temperature, branching and chemical structure. It also scales like l_c \propto n.
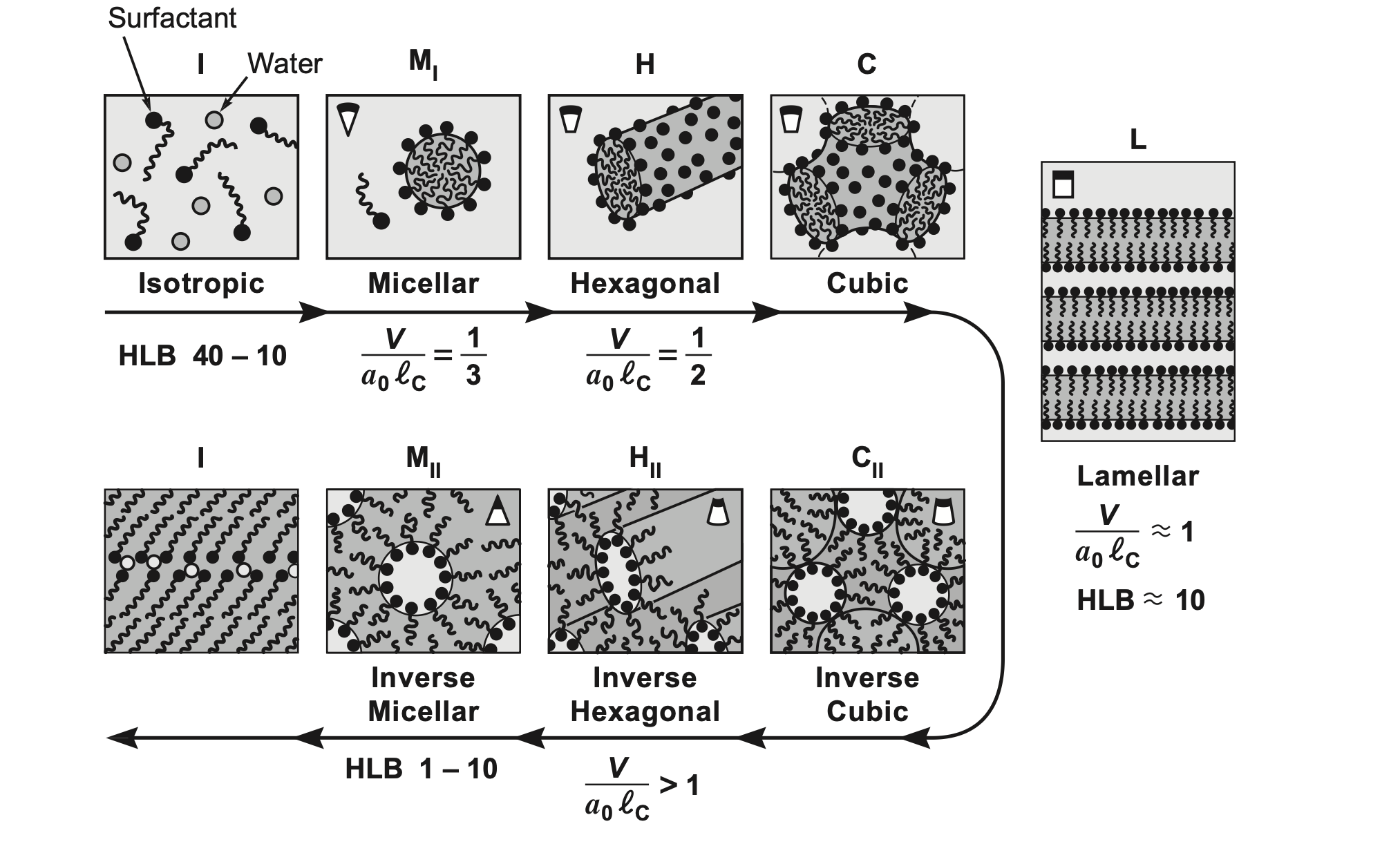
A balance between energy and entropy leads to different self-assembled structures .
Different types of self assembled structures: (A) spherical micelles, (B) cylindrical micelles and (C) bilayers.
Spherical micelle
For a spherical micelle of radius R with aggregation number N , the total volume and surface area are given by
\begin{gathered}
N v=\frac{4 \pi}{3} R^{3} \\
N a_{0}=4 \pi R^{2}\end{gathered}
where we used the fact that the radius R cannot be larger than the critical chain length \mathrm{l}_{\mathrm{c}}. We thus obtain for the critical packing parameter P
P=\frac{v}{a_{0} l_{c}}<\frac{1}{3}
Cylindrical micelles
For a cylindrical micelle of length L the total volume and surface area are given by N v=\pi R^{2} L
and
N a_{0}=2 \pi R L
Therefore \dfrac{v}{a_{0}}=\dfrac{R}{2}<\dfrac{l_{c}}{2} again using R<l_{C}.
We thus obtain for the critical packing parameter P=\dfrac{v}{a_{0} l_{c}} that \dfrac{1}{3}<P<\dfrac{1}{2}
Below the lower limit spherical micelles are formed.
Bilayers (lamellar)
For a bilayer (with separation D between the layers and area A) the total volume and surface area are given by
\begin{aligned}
& N v=A D \\
& N a_{0}=2 A \\
& \frac{v}{a_{0}}=\frac{D}{2}<l_{c} \quad\left(\text { using } D<2 l_{c}\right)
\end{aligned}
We thus obtain for the critical packing parameter P=\dfrac{v}{a_{0} l_{c}} that \dfrac{1}{2}<P<1
Below the lower limit cylindrical micelles are formed.
Packing parameter summary
Mesophases formed by surfactants as we vary the packing parameter P=\dfrac{v}{a_{0} l_{c}} , from Israelachvili, J. N., Intermolecular and Surface Forces, 3rd Edition, Academic Press, 2011.